Cada planta e animal se caracteriza por um conjunto de cromossomos, representado uma vez em células haploides (por exemplo gametas e esporos) e duas vezes em células diploides .
Cada espécie tem um número específico de cromossomos. Mas, as vezes ocorre irregularidades na divisão nuclear, ou podem acontecer "acidentes" (como os de radiação) nos cromossomos de interfase de modo que se podem formar células ou organismos inteiros com genomas aberrantes. Tais aberrações cromossômicas podem incluir genomas inteiras, cromossomos isolados inteiros, ou só partes de cromossomos. As aberrações cromossômicas podem ser numéricas ou estruturais e envolver um ou mais autossomos, cromossomos sexuais ou ambos. As aberrações cromossômicas numéricas incluem os casos em que há aumento ou diminuição do número do cariótipo normal da espécie humana, enquanto as aberrações cromossômicas estruturais incluem os casos em que um ou mais cromossomos apresentam alterações de sua estrutura Assim, os citologistas reconhecem:
(1)Alterações no Número de Cromossomos (Heteroploidia)
(2)Alterações na Estrutura dos Cromossomos.
A heteroploidia pode atingir conjuntos inteiros de cromossomos (euploidia) ou perda ou adição de cromossomos inteiros isolados (aneuploidia). Todas essas alterações tem um importante efeito sobre o desenvolvimento, pois ao alterar a estrutura nuclear normal podem produzir alterações fenotípicas.
Alterações no número de cromossomos
As aneuploidias devem-se à não separação (ou não-segregação) de um (ou mais) cromossomo(s) para as células-filhas durante a meiose ou durante as mitoses do zigoto A não-segregação na mitose decorre do não-rompimento do centrômero no início da anáfase ou da perda de algum cromossomo por não ter ele se ligado ao fuso.
A não-segregação na meiose é devida a falhas na separação dos cromossomos ou das cromátides, que se separam ao acaso para um polo ou outro. Na meiose a não-segregação tanto pode ocorrer na primeira divisão como na Segunda. No primeiro caso, o gameta com o cromossomo em excesso, em lugar de ter apenas um dos cromossomos de um dado par, ou seja, terá um cromossomo paterno e um materno. No segundo, o gameta com o cromossomo em excesso terá dois cromossomos paternos ou dois maternos.
Quando em consequência desses processos de não-segregação falta um cromossomo de um dado par, isto é, quando o número de cromossomos da célula é 2n - 1, diz-se, que a célula apresenta monossomia para este cromossomo. Se faltam os dois elementos do mesmo par 2n - 2, tem-se nulisomia. Se, pelo contrário, houver aumento do número de cromossomos de um determinado par, a célula será polissômica para o cromossomo em questão; ela será trissômica, tetrassômica, pentassômica etc., conforme tiver 1, 2 ou 3 cromossomos a mais, sendo, nesses casos, o seu número cromossômico designado por (2n + 1), (2n + 2), (2n + 3) etc.
Euplodias
As euploidias consistem na variação numérica do conjunto básico de cromossomos, designado n. Compreendem a haploidia e a poliploidia.
♦ Monoploidia ou haploidia
Na maioria das algas e fungos e em todas as briófitas, a fase haplóide representa a fase predominante do ciclo vital.
Naturalmente não há dominância e recessividade nos haploides enquanto a gametogênese é desprovida da meiose.
♦ Poliploidia
É a existência de três ou mais conjuntos cromossômicos básicos nas células. Tais organismos são designados triploides (3n), tetraploides (4n), pentapresidentes (5n) etc.
Os poliploides podem ser autopoliploides e alopoliploides.
Nos autopoliploides existem três ou mais conjuntos cromossômicos básicos (genomas) da mesma espécie.
Na drosófila, por exemplo, a fêmea pode produzir óvulos não-reduzidos, isto é, diplóides (2n) que, fertilizados, originam fêmeas triploides (3n).
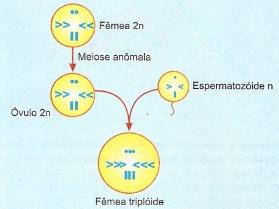
Aberrações por poliploidia em drosófila
Os alopoliploides originam-se da duplicação dos genomas de um híbrido diploide resultante de cruzamento interespecífico.
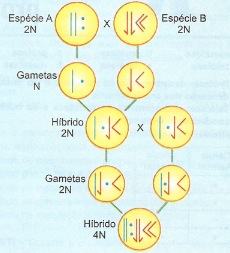
O Crruzamento interespecífico
O exemplo clássico de alopoliploide experimental é o vegetal Raphanobrassica, estudado por Karpechenko.
O rabanete Raphanus sativus e a couve Brassica oleracea são espécies diferentes, mas apresentam o mesmo número de cromossomos, ou seja, 18. Quando cruzadas, podem originar um híbrido interespecífico de grande vigor, mas estéril por apresentar 2 genomas diferentes. Contudo, formam-se alguns gametas não-reduzidos, contendo 18 cromossomos.
Da união de tais gametas originam-se zigotos viáveis, produzindo sementes com 36 cromossomos, sendo 18 de Raphanus e 18 de Brassica. Tais sementes podem produzir plantas tetraploides férteis, constituindo uma nova espécie, a Raphanobrassica.
O rabanete Raphanus sativus e a couve Brassica oleracea são espécies diferentes, mas apresentam o mesmo número de cromossomos, ou seja, 18. Quando cruzadas, podem originar um híbrido interespecífico de grande vigor, mas estéril por apresentar 2 genomas diferentes. Contudo, formam-se alguns gametas não-reduzidos, contendo 18 cromossomos.
Da união de tais gametas originam-se zigotos viáveis, produzindo sementes com 36 cromossomos, sendo 18 de Raphanus e 18 de Brassica. Tais sementes podem produzir plantas tetraploides férteis, constituindo uma nova espécie, a Raphanobrassica.
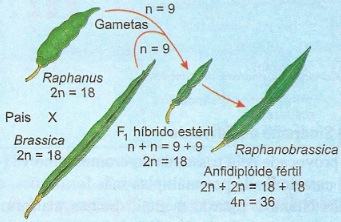
Origem da Raphanobrassica
Examinando-se o grande número de formas poliploides existentes, principalmente nos vegetais, pode-se concluir que há uma relação entre a multiplicação do número de cromossomos e a evolução desses organismos.
Uma das formas da poliploidia, a alopoliploidia, deve ter contribuído bastante na formação de novas espécies, que puderam ser obtidas experimentalmente pela anfidiploidia, isto é, reunião de 2 genomas diferentes num indivíduo e a subseqüente duplicação do número de cromossomos. O fumo, por exemplo, originou-se do cruzamento de duas espécies distintas, com duplicação do número de cromossomos. As espécies que contribuíram para a formação dessa planta são Nicotiana süvestris e Nicotiana tomentosiformes, ambas com 12 pares de cromossomos, obviamente diferentes. O híbrido é a espécie Nicotiana tabacum.
Na espécie humana a poliploidia, como era de se esperar, raramente acontece e os casos relatados são de abortos espontâneos ou natimortos.
Aneuploidias dos cromossomos sexuais
Síndrome devida a uma aberração cromossômica, caracterizada por fenótipo feminino, nanismo, infantilismo genital, disgenesia das gônadas (ovários reduzidos) e malformações diversas. O cariótipo é 45, X em 60% dos casos. As meninas com esta síndrome são identificadas ao nascimento ou antes da puberdade por suas características fenotípicas distintivas.
As anormalidades envolvem baixa estatura, disgenesia gonadal, pescoço alado, tórax largo com mamilos amplamente espaçados e uma frequência elevada de anomalias renais e cardiovasculares.
Síndrome de Klinefelter 47-XXY
Síndrome que associa no homem jovem um desenvolvimento anormal dos seios (ginecomastia), atrofia testicular, ausência de formação de espermatozoides (azoospermia) e uma elevação da concentração do hormônio hipofisário FSH. (O cariótipo mais comum é 47, XXY. Na sua forma habitual, porém não de regra, a síndrome de Klinefelter pode ser acompanhada de deficiência intelectual. As demais variantes cromossômicas são as responsáveis por uma debilidade mental bem mais grave). A Síndrome caracteriza-se pela presença do cariótipo 47, XXY ou em mosaicos.
Os pacientes são altos e magros, com membros inferiores relativamente longos. Após a puberdade os sinais de hipogonadismo se tornam óbvios. Os testículos permanecem pequenos e os caracteres sexuais secundários continuam subdesenvolvidos.

Aneuploidias Autossômicas
Síndrome de Down
Doença congênita caracterizada por malformações dos órgãos (coração, rins), retardamento mental de moderado a severo, língua espessa, pés e mãos de pequenas dimensões, alterações nas feições. É resultante de uma anormalidade na constituição cromossômica: os indivíduos afetados apresentam um cromossomo extra - que se acrescenta ao par de número 21 - em suas células (por esta razão a doença é também denominada trissomia do 21). O termo mongolismo é um sinônimo usual: a presença de fendas palpebrais oblíquas faz lembrar os indivíduos das raças orientais. A frequência com que esta síndrome se manifesta é de uma para cada 500 crianças nascidas vivas e é superior para concepções em mulheres com idade acima de 40 anos. Esta síndrome foi descrita em 1866 pelo médico inglês John Langdon Haydon Down (1828 - 1896). A Síndrome de Down ou trissomia do 21, é sem dúvida o distúrbio cromossômico mais comum e a mais comum forma de deficiência mental congênita. Geralmente pode ser diagnosticada ao nascimento ou logo depois por suas características dismórficas, que variam entre os pacientes, mas produzem um fenótipo distintivo.
Os pacientes apresentam baixa estatura e o crânio apresenta braquicefalia, com o occipital achatado. O pavilhão das orelhas é pequeno e dismórfico. A face é achatada e arredondada, os olhos mostram fendas palpebrais e exibem manchas de Brushfield ao redor da margem da íris. A boca é aberta, muitas vezes mostrando a língua sulcada e saliente. As mãos são curtas e largas, frequentemente com uma única prega palmar transversa ("prega simiesca").
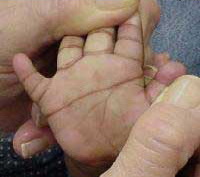
Mão de uma criança com Síndrome de Down.
Trissomia do 13 -Patau
A trissomia do 13 é clinicamente grave e letal em quase todos os casos que sobrevivem até 6 meses de idade. O cromossomo extra provém de não-disjunção da meiose I materna e cerca de 20% dos casos resultam de uma translocação não-balanceada.
O fenótipo inclui malformações graves do sistema nervoso central como arrinencefalia. Um retardamento mental acentuado está presente. Em geral há defeitos cardíacos congênitos e defeitos urigenitais. Com frequência encontram-se fendas labial e palatina, anormalidades oculares, polidactilia, punhos cerrados e as plantas arqueadas.

Fenda Labial
Trissomia do 18-Edwards
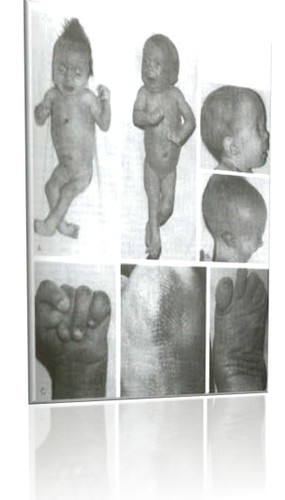
Foi descrita primeiramente em 1960, por John H. Edwards, em recém-nascidos que apresentavam malformações congênitas múltiplas e retardamento mental. Esta foi a segunda síndrome revelada no homem, sendo que a primeira foi a síndrome de Down ou trissomia 21.
Acomete 1 em cada 8.000 nascidos, sendo o sexo feminino mais comumente afetado. Entretanto, acredita-se que 95% dos casos dessa síndrome resultem em aborto espontâneo durante a gestação. A expectativa de vida para um portador da síndrome de Edwards é baixa; todavia, já foram descritos casos de adolescentes com 15 anos de idade portadores da afecção.
A maior parte dos pacientes portadores dessa síndrome apresenta trissomia regular sem mosaicismo, ou seja, cariótipo 47, XX ou XY, +18. Dentre os restantes, aproximadamente metade é formada por casos de mosaicismo e outra parcela por problemas mais complexos, como aneuploidias duplas, translocações. Destes, cerca de 80% dos casos são resultantes de uma translocação abrangendo todo ou quase todo o cromossomo 18, sendo que este pode ser recebido ou adquirido novamente a partir de um progenitor transportador.
As características apresentadas pelos portadores da trissomia 18 são retardamento físico, choro fraco, hipotonia seguida de hipertonia, hipoplasia da musculatura esquelética e do tecido adiposo subcutâneo, redução de resposta a estímulos sonoros, retardo mental e diversas características físicas, como:
Crânio disfórmico;
Face triangular com testa alta e plana;
Maxilares recuados;
Orelhas mal formadas e baixas;
Occipital proeminente;
Lábio leporino e/ou fenda palatina;
Pescoço curto com pelos em excesso;
Externo curto;
Mamilos pequenos;
Presença de hérnia inguinal ou umbilical;
Manutenção dos punhos cerrados é característico;
Pé torto congênito é comum;
Encurtamento do hálux (dedão do pé);
Rugas nas palmas das mãos e plantas dos pés;
Nos meninos é comum a ocorrência de criptorquidia, já nas meninas é comum a hipertrofia de clitóris com hipoplasia dos grandes lábios;
Diversas malformações congênitas podem ser encontradas, afetando o cérebro, coração, rins e aparelho gastrointestinal. Entre as malformações cardíacas mais frequentes, que normalmente é a causa do óbito nesses pacientes, está a comunicação interventricular e a persistência do ducto arterial. Também observa-se com frequência a presença de tecido pancreático heterotrópico, eventração diafragmática, divertículo de Meckel e diferentes tipos de displasias renais.
Ainda dentro da barriga, já é possível detectar e presença de anomalias nos fetos. O exame ultra-sonográfico transvaginal, entre 10 a 14 semanas de gestação, possibilita estimar a espessura do “espaço escuro” existente entre a pele e o tecido subcutâneo, que reveste a coluna cervical fetal, detectando, deste modo, alterações no feto.
O diagnóstico diferencial deve ser feito com a síndrome da trissomia 13 (ou síndrome de Patau), pois em ambas os indivíduos podem apresentar lábio leporino e/ou fenda palatina.
Quando há o aparecimento dessa síndrome, aconselha-se procurar aconselhamento genético, para que seja realizado um estudo genético.
O prognóstico para indivíduos que nascem com essa doença genética é ruim, sendo a sobrevida da maioria desses pacientes é de 2 a 3 meses para os meninos e 10 meses para as meninas, muito dificilmente ultrapassando os 2 anos de vida; os pacientes que possuem o mosaicismo podem sobreviver por mais tempo.
Síndrome do Duplo Y - 47-XYY

A síndrome XYY ou síndrome de Jacobs é uma aneuploidia dos cromossomas sexuais, onde um humano do sexo masculino recebe um cromossoma Y extra em cada célula, ficando assim com um cariótipo 47,XYY. A síndrome XYY também é designada como trissomia XYY, aneuploidia 47,XYY ou síndrome do super-macho.
- A maioria dos Homens com XYY são fenotipicamente normais
- Crescimento ligeiramente acelerado na Infância
- Homens com estatura muito elevada
- antigamente associada a comportamento anti-social, porém são relatados problemas comportamentais como distração, hiperatividade e crises de fúria na infância e início da adolescência, sendo que o comportamento agressivo usualmente não é problema e eles aprendem a controlar a raiva à medida que crescem.
- Ocorrência 1/1.000 nascimentos do sexo masculino.
Causas
O cromossomo Y transporta relativamente poucos genes, por esse motivo essa síndrome não apresenta tantas anomalias físicas. Essa síndrome também é provocada quando a idade materna é avançada. Não há fatores claramente definidos predispondo à ocorrência de 47, XYY. No entanto, foi constatado que o fumo na adolescência está associado a um aumento na dissomia presente no esperma e uma diminuição em aspectos específicos da qualidade do sêmen. Isso pode afetar a fertilidade no homem e pode também aumentar a chance de aneuploidia na prole, predispondo a ocorrência de 47, XYY. Além disto, o cromossomo Y é muito característico em espécies de protozoários, sendo considerada uma raridade pelos protozoários não terem material genético totalmente estruturado! Os humanos, por exemplo, possuem 46 cromossomos: 23 do pai (esperma) e 23 da mãe (óvulo).
Trissomia do Triplo-X 47-XXX

Algumas mulheres podem apresentar até 4 ou 5 cromossomos X extras. Quanto mais cromossomos X, maior o índice de retardo mental nessas mulheres.
Existem mulheres portadoras dessas aberrações cromossômicas, mas desconhecem a doença por não apresentarem os sintomas.
Os sintomas dessa síndrome envolvem:
- Menor grau de inteligência
- Pessoa com características sexuais e comportamentais femininas
- Mulheres tendem a ser altas
- Múltiplas peles frouxas no pescoço
- São férteis
- Podem entrar na menopausa precocemente
- Retardamento mental (o grau de retardamento varia entre as portadoras)
Algumas portadoras são férteis, mas em alguns casos a doença pode causar esterilidade.
Mulheres portadoras podem dar origem a crianças perfeitamente normais.
As crianças que nascem com essa síndrome não apresentam claramente os sintomas logo após o nascimento, mas podem apresentar baixo peso.
A doença pode ser diagnosticada por estudos do cariótipo, onde é claramente visível a presença de três cromossomos sexuais X.
Alterações na estrutura dos cromossomos
São alterações que não modificam a quantidade de cromossomos de uma célula, mas determinam o aparecimento de cromossomos anormais. As aberrações que vamos descrever a seguir quase sempre implicam em problemas sérios , inclusive na formação de gametas . Isso porque durante a meiose, o cromossomo com a deficiência pareia de forma anômola com seu homólogo que não sofreu alteração, afetando o andamento , do processo meiótico. A gravidade das manifestações de uma deficiência depende dos genes ausentes. Um exemplo humano é a sindrome de cri du chat, em que falta um fragmento do braço curto do cromossomo 5. Outro exemplo é o cromossomo 22 curto ("cromossomo Filadélfia"), associado a uma forma de leucemia. As duplicações são menos graves que as deficiências porque não provocam falta de informações genéticas. Exemplos, a seguir:
Deficiência ou Deleção
É a perda de uma parte do cromossomo. Segundo a posição, a deficiência pode ser terminal ou intercalar. As terminais são mais raras que as intercalares. Os fragmentos que permanecem com o centrômero passam a funcionar como cromossomos deficientes. Os pedaços que não contiverem o centrômero irão perder-se, pois, na mitose, eles não podem prender-se às fibras do fuso. As deficiências representam perda de genes e só permitirão a sobrevivência se forem diminutas, ou incidirem em material cromossômico de pouca importância. A deficiência pode ser originada de uma volta, seguida de uma ruptura, como mostra a figura abaixo.
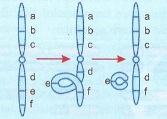
A formação de uma deleção
Em drosófila é bem conhecida a alteração Notch, uma aberração das asas produzida por uma deficiência no cromossomo X, constituindo um caráter sex-linked. A deficiência também atinge o lócus contendo os genes V para olho vermelho e v para olho branco. A deficiência Notch é sempre letal nos machos; nas fêmeas só é letal quando homozigótica. A figura abaixo ilustra a herança da deficiência Notch através do cruzamento de uma fêmea Notch de olho vermelho com um macho normal de olho branco.
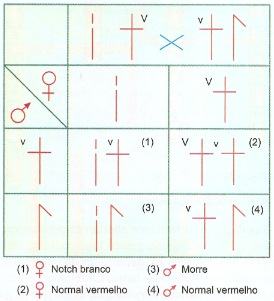
Na mulher, a perda de parte de um cromossomo X não é letal porque, de qualquer modo, um dos cromossomos X das fêmeas dos mamíferos é praticamente inativo.
No homem, existe um caso de deficiência autossômica que determina a leucemia mielóide crônica, uma variedade de câncer. Os pacientes têm 46 cromossomos, mas um dos que pertencem ao grupo 22 é menor. Tal cromossomo é designado como cromossomo de Philadelphia.
Uma deficiência do cromossomo número 5 causa, no homem, a síndrome do miado de gato, assim chamada em virtude do choro dos afetados lembrar um gato miando. A síndrome mostra retardamentos neuromotor e mental graves, bem como hipotrofia muscular.
Síndrome do miado de gato
Caracterizada por retardo mental, microcefalia, aspecto arredondado da face, presença de dobras epicânticas nos olhos e de choro semelhante a um miado de gato. (É provocada por uma deleção do braço curto do cromossomo 5.)
Inversão
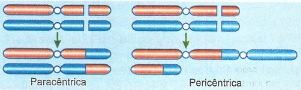
A inversão consiste em duas fraturas cromossômicas seguidas da reconstituição com o pedaço invertido entre as mesmas. Se ocorre em um único braço do cromossomo é chamada de paracêntrica; se envolve o centrômero, ela é pericêntrica.
 Tipos de inversão
Tipos de inversão
Translocação
A translocação é a transferência de parte de um cromossomo para um cromossomo não-homólogo.
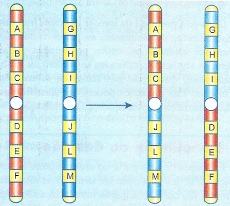
Aberração por translocação
Pode ser paracêntrica e pericêntrica.
A quebra de um cromossomo produz um fragmento sem centrômero (acêntrico) e outro com centrômero (cêntrico). Na translocação paracêntrica cada fragmento acêntrico se solda a um fragmento cêntrico, recompondo dois cromossomos que têm centrômero. Na translocação pericêntrica, os fragmentos cêntricos se soldam um ao outro e os acêntricos também se juntam: um dos cromossomos translocados fica dicêntrico (com 2 centrômeros) e outro acêntrico.
Tipos de translocação
O cromossomo acêntrico tende a perder-se e o dicêntrico tende a romper-se, pois na anáfase os dois centrômeros podem migrar para os pólos opostos. Por isso, é raro encontrar nas células os produtos das translocações pericêntricas, enquanto os resultados das translocações paracêntricas podem persistir em todas as células do organismo, como já se encontraram na espécie humana.
Os isocromossomos são cromossomos que mostram, simultaneamente, um braço com deficiência total e outro com duplicação completa. Tais cromossomos aparecem por divisão longitudinal e não transversal do centrômetro.
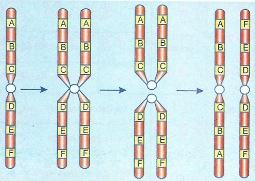
Formação de isocromossomos
Diagnóstico das Anomalias :
Amniocentese
Punção da cavidade amniótica através da parede abdominal, feita numa mulher grávida; permite a retirada de certa quantidade de líquido amniótico para fins de análise. A amniocentese precoce, praticada entre 16° e 18° semana de gestação, permite fazer o diagnóstico de anomalias fetais que podem conduzir a um aborto; também é possível detectar se a criança é portadora de mongolismo, anencefalia ou outra anormalidade genética. A amniocentese tardia, feita no terceiro trimestre da gravidez, serve para evidenciar se há sofrimento fetal crônico.
Na herança autossômica dominante um fenótipo é expressado da mesma maneira em homozigotos e heterozigotos. Toda pessoa afetada em um heredograma possui um genitor afetado, que por sua vez possui um genitor
Nos casamentos que produzem filhos com uma doença autossômica dominante, um genitor geralmente é heterozigotico para a mutação e o outro genitor é homozigótico para o alelo normal. Pode-se escrever os genótipos dos pais como: Cada filho desse casamento tem uma chance de 50% de receber o alelo anormal (A) do genitor afetado e, portanto ser afetado (A/a), e uma chance de 50% de receber o alelo normal (a) e, assim não ser afetado (a/a).
Critérios da Herança Autossômica Dominante
1 - O fenótipo aparece em todas as gerações, e toda pessoa afetada tem um genitor afetado.
2 - Qualquer filho de genitor afetado tem um risco de 50% de herdar o fenótipo.
3 - Familiares fenotipicamente normais não transmitem o fenótipo para seus filhos.
4 - Homens e Mulheres têm a mesma probabilidade de transmitir o fenótipo aos filhos de ambos os sexos.
Exemplos de Herança Autossômica Dominante2 - Qualquer filho de genitor afetado tem um risco de 50% de herdar o fenótipo.
3 - Familiares fenotipicamente normais não transmitem o fenótipo para seus filhos.
4 - Homens e Mulheres têm a mesma probabilidade de transmitir o fenótipo aos filhos de ambos os sexos.
Doença de Huntington (DHq) É uma doença neurodegenerativa fatal de herança autossômica dominante caracterizada por movimentos involutários e demência progressiva. O aparecimento da doença se dá entre os 30-50 anos de idade sendo 38 a idade média de aparecimento. O gene foi mapeado no cromossomo 4p16 em 1981 por técnicas de genética molecular. É possível identificar os indivíduos portadores do gene.
Neurofibromatose de Von Recklinghausen (NF1) É um distúrbio comum do sistema nervoso caracterizado por manchas café-com-leite e tumores cutâneos fibromatosos.
Herança Autossômica Recessiva
Os distúrbios autossômicos recessivos expressam-se apenas em homozigotos, que, portanto, devem ter herdado um alelo mutante de cada genitor. Desse modo:
O risco de seus filhos receberem o alelo recessivo de cada genitor, e serem afetados é de 1/4. A maioria dos genes dos distúrbios autossômicos recessivos está presente em portadores dos genes. Eles podem ser transmitidos nas famílias por numerosas gerações sem jamais aparecer na forma homozigótica. A chance de isto acontecer é aumentada se os pais forem aparentados. A consanguinidade dos genitores de um paciente com um distúrbio genético é uma forte evidência em favor da herança autossômica recessiva daquela afecção.
Critérios da Herança Autossômica Recessiva
1 - O fenótipo é encontrado tipicamente apenas na irmandade do probando e o fenótipo salta gerações.
2 - O risco de recorrência para cada irmão do probando é de 1 em 4.
3 - Os pais do indivíduo afetados em alguns casos são consanguíneos.
4 - Ambos os sexos têm a mesma probabilidade se serem afetados.
2 - O risco de recorrência para cada irmão do probando é de 1 em 4.
3 - Os pais do indivíduo afetados em alguns casos são consanguíneos.
4 - Ambos os sexos têm a mesma probabilidade se serem afetados.
Exemplos de Herança Autossômica Recessiva
Fibrose Cística
Doença autossômica recessiva caracterizada por doença pulmonar crônica, insuficiência pancreática exócrina, aumento da concentração de cloreto no suor. O defeito básico é uma mutação do gene que codifica a proteína reguladora da fibrose cística, provavelmente envolvida no transporte de ânions através da membrana celular.
Doença de TAY-SACHS
Distúrbio neurológico degenerativo, autossômico recessivo, que se desenvolve quando a criança tem 6 meses de idade. Há uma deterioração mental e física intensa desde a lactância, a morte ocorre entre 2 e 3 anos de idade. O defeito básico são mutações no lócus da subunidade a da hexosaminidase A. A deficiência ou ausência da subunidade a da hexosaminidase A leva ao acúmulo do gangliosídeo GM2, principalmente nos neurônios.
Mutações Gênicas
Em 1941, os pesquisadores Beadle e Tatum, fazendo experiências com um tipo de bolor de pão, a Neurospora sp, observaram que nem sempre a autoduplicação do DNA ocorria de modo perfeito. O bolor crescia num meio de cultura contendo açúcar e diversos sais inorgânicos. Seus esporos eram submetidos a raios X e alguns deles passavam depois a produzir bolores com novas características. Por exemplo, alguns perdiam a capacidade de fabricar lisina e só conseguiam sobreviver quando aquele aminoácido era acrescentado ao meio de cultura. Essa incapacidade foi relaciona com a falta de uma enzima necessária para a síntese de lisina. Concluíram, então, que os raios X teriam danificado a formação daquele tipo específico de enzima.
Como a produção de uma enzima depende de informação codificada no DNA, a conclusão daqueles pesquisadores ficou conhecida como a relação "um gene - uma enzima". Atualmente, fala-se, com maior precisão, na relação "um gene - uma cadeia polipeptídica".
A modificação genética induzida através dos raios X é conhecida como mutação. As mutações podem resultar de uma alteração na sequência dos nucleotídeos, ou de quebras e mudanças de posição dos fragmentos da molécula de DNA. Portanto são mutações as alterações numéricas e estruturais dos cromossomos, que persistem através das autoduplicações, transmitindo-se às células-filhas. Existem também erros que ocorrem no RNA, no momento das transcrições ou das traduções, e afetam somente a própria célula.
Agentes Mutagênicos
As mutações são produzidas por agentes mutagênicos, que compreendem principalmente vários tipos de radiação, dentre os quais os raios ultravioleta, os raios X e substâncias que interferem na autoduplicação do DNA ou na transcrição do RNAm, determinando erros nas sequências dos nucleotídeos. Os agentes mutagênicos são fatores que podem elevar a frequência das mutações. Em 1920, Hermann J. Muller descobriu quem submetendo drosófilas ao raio X, a frequência das mutações aumentava cerda de cem vezes em relação à população não exposta. O aumento na taxa de mutações pode ser obtido pelo emprego de numerosos agentes físicos e químicos.
A lista das substâncias mutagênicas tem aumentado muito nos últimos anos, sendo bastante conhecidos o gás mostarda, o ácido nitroso, a bromouracila, o formaldeído, a nicotina. Vários tipos de câncer podem ser produzidos por alterações ocorridas nos ácido nucléicos; por isso os mesmos agentes mutagênicos podem ser também cancerígenos. Porém, a mais importante dentre eles são as radiações. Quando uma célula recebe radiação, as moléculas podem ser quebradas ou alteradas em suas estruturas. Quando as alterações são muito grandes, podem interferir com o metabolismo e divisão celular, e a célula morre. Quando ela sobrevive à radiação, as modificações são duplicadas e transmitidas para as células das gerações sucessivas.
Entre os agentes físicos, os mais conhecidos são as radiações, bem como o raio X. O calor também aumente a incidência das mutações: na espécie humana, sua frequência em trabalhadores de altos-fornos de usinas siderúrgicas, os quais permanecem muito tempo em locais de temperatura elevada, é mais alta que na população geral.
Todos os seres vivos estão submetidos, diariamente, a vários desses agentes. Entretanto, as mutações permanecem como eventos não muito frequentes. A relativa estabilidade do material genético deve-se à existência de um grupo de enzimas de reparação, que "patrulham" permanentemente as moléculas de DNA à caça de alterações na sequência de seus nucleotídeos. Na maioria das vezes, essas alterações são detectadas e consertadas.
Síndrome de Turner
A Síndrome de Turner é uma monossomia do X e apenas 1% dos que a possuem sobrevive. A proporção estatística é de 1 para 8000 nascimentos, sendo que apenas a metade das meninas que sobrevivem apresentam cariótipo com 45X (como no cariótipo abaixo), a outra metade tem muitas anormalidades cromossômicas no cromossomo sexual.

Cariótipo de uma pessoa com Síndrome de Turner
 Com tamanha alteração não é de se espantar que a maioria nem chegue a nascer, e das que nascem apenas uma pequena parcela sobrevive. E dentre as que sobrevivem, aproximadamente 90% precisam fazer uma substituição hormonal para auxiliar no desenvolvimento dos caracteres sexuais secundários (pêlos pubianos, por exemplo) já que o organismo não consegue fazê-lo sozinho. Por esses dados é possível confirmar o que as pesquisas já dizem: a causa mais frequente dos abortos espontâneos por síndromes é a Síndrome de Turner, dados mais específicos taxam em 18% este índice.
Com tamanha alteração não é de se espantar que a maioria nem chegue a nascer, e das que nascem apenas uma pequena parcela sobrevive. E dentre as que sobrevivem, aproximadamente 90% precisam fazer uma substituição hormonal para auxiliar no desenvolvimento dos caracteres sexuais secundários (pêlos pubianos, por exemplo) já que o organismo não consegue fazê-lo sozinho. Por esses dados é possível confirmar o que as pesquisas já dizem: a causa mais frequente dos abortos espontâneos por síndromes é a Síndrome de Turner, dados mais específicos taxam em 18% este índice.
A síndrome de Turner ocorre graças à um erro durante a gametogênese, causando a monossomia do X e quase sempre está presente no gameta paterno. Suas características fenotípicas mais comuns são: estatura menor que o padrão para a idade, pescoço robusto (ou alado), ausência da maturação sexual, tórax largo com mamilos muito separados, inchaço nas mãos e pés. Nas figuras abaixo e ao lado podemos perceber perfeitamente este padrão fenotípico.

Graças às características observadas acima, erroneamente no passado as mulheres acometidas por esta síndrome eram incluídas no grupo dos deficientes mentais, pela semelhança de alguns caracteres. Mas isto é um equívoco, pois estudos e relatos de caso já comprovaram que ou a inteligência não é afetada ou comumente as mulheres portadoras desta síndrome evoluem sua inteligência um pouco mais que a média das outras mulheres, ditas “normais” (ou não-sindromizadas).
Síndrome de Klinefelter
A síndrome de Klinefelter trata-se de uma anomalia (aneuploidia), uma mutação cromossômica numérica, há acréscimo de um cromossomo sexual no conjunto diploide de um indivíduo.
Essa alteração quantitativa foi inicialmente descrita por volta de 1940, pelo cientista Harry Klinefelter, na ocasião em que analisava a demanda de oxigênio pela glândula adrenal, deparando-se com um caso raro de ginecomastia, evidenciado através do proeminente desenvolvimento das mamas (dos seios) em indivíduo do gênero masculino.
Entre as características constatadas, a baixa estatura, a partir de exames o Dr. Klinefelter observou uma variação irregular nos níveis endócrinos esperados para um organismo do sexo masculino, registrando uma elevada síntese de hormônio gonadotrópico (GnRH), folículo estimulantes (FSH) e luteinizante (LH).
Essa alteração quantitativa foi inicialmente descrita por volta de 1940, pelo cientista Harry Klinefelter, na ocasião em que analisava a demanda de oxigênio pela glândula adrenal, deparando-se com um caso raro de ginecomastia, evidenciado através do proeminente desenvolvimento das mamas (dos seios) em indivíduo do gênero masculino.
Entre as características constatadas, a baixa estatura, a partir de exames o Dr. Klinefelter observou uma variação irregular nos níveis endócrinos esperados para um organismo do sexo masculino, registrando uma elevada síntese de hormônio gonadotrópico (GnRH), folículo estimulantes (FSH) e luteinizante (LH).

Aparência anatômica do seio e do aparelho reprodutor de um homem com Síndrome de Klinefelter.
Correlacionou então tais resultados à capacidade reprodutiva, onde verificou sinais clínicos como: testículos pequenos e esterilidade (infertilidade) por azoospermia, situação de ausência completa de espermatozoides no sêmen.
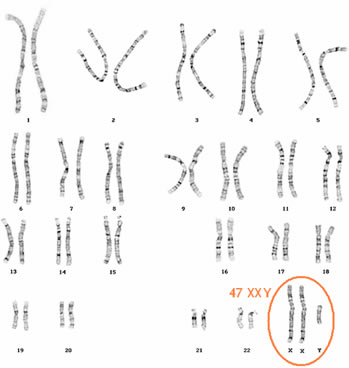
Ideograma (cariótipo: 47,XXY), relativo à Síndrome de Klinefelter.
A frequência dessa síndrome com um cromossomo X extra, acomete um menino em aproximadamente 850 nascimentos.
Fontes: http://www.algosobre.com.br/biologia/aberracoes-cromosomicas.html
http://www.coladaweb.com/biologia/genetica/aberracoes-cromossomicas
Questões sobre aberrações cromossômicas
1) Uma aneuploidia freqüente na espécie humana é a síndrome de Down, também chamada de mongolismo, que se caracteriza pela:
a) não-disjunção de cromossomos sexuais durante a gametogênese;
b) ausência de um cromossomo sexual;
c) trissomia do cromossomo 21;
d) monossomia do cromossomo 21;
e) trissomia de um cromossomo sexual.
2) (UF - Ceará) Qual das seguintes síndromes humanas é devida a uma monossomia?
a) Síndrome de Down
b) Síndrome de Turner
c) Síndrome de Klinefelter
d) Síndrome de Kernicterus
e) Síndrome da imunodeficiência adquirida.
3) Qual a origem dos organismos aneuplóides?
4) O gênero vegetal Spartina é composto de várias espécies que apresentam o seguinte número de cromossomos em suas células meristemáticas:
I. S. stricta – 56
II. S. alterniflora – 70
III. S. townsendii – 126
A espécie S. townsendii é, provavelmente:
a) um poliplóide de I;
b) um aneuplóide de II;
c) descendente de espécie diferente de I e II;
d) apenas um gíbrido de I e II;
e) um híbrido poliplóide de I e II.
5) Qual é a conseqüência da Síndrome de Turner:
a) Desvios de personalidade;
b) Alta taxa de hormônios estrógenos;
c) Apresenta deficiência mental;
d) Baixa taxa de hormônios estrógenos.
6) Qual é a conseqüência da Síndrome de Klinefelter:
a) Infertilidade feminina;
b) Fertilidade feminina;
c) Infertilidade masculina;
d) Fertilidade masculina.
7) Das alternativas abaixo qual não é consequência da Síndrome de Down?
a) Baixa estrutura e magro.
b) Microcefalia.
c) Deficiências motoras.
d) Problemas no coração e na audição.
8) (UVA) Sobre os portadores de Síndrome de Down ou mongolismo são corretas as afirmações, exceto:
a) Possuem alterações do tipo mutações cromossômicas autossômicas.
b) Possuem um cromossomo a mais no par 21.
c) Possuem baixo coeficiente intelectual.
d) Possuem mutações relacionadas aos cromossomos sexuais.
9) Responda esta questão com base no esquema abaixo, que representa os cromossomos sexuais da espécie humana.
 Os genes que determinam o daltonismo e a hemofilia devem estar situados:
Os genes que determinam o daltonismo e a hemofilia devem estar situados:
a) Apenas no segmento II.
b) Nos segmentos II e I, respectivamente.
c) Apenas no segmento I.
d) Nos segmentos I e II, respectivamente.
e) Nos segmentos I ou II, indiferentemente.
10) Responda esta questão com base na figura abaixo. Nela, A representa uma célula com 2n cromossomos. Os esquemas B e C representam, respectivamente:
 a) Monossomia e trissomia.
a) Monossomia e trissomia.
b) Haploidia e trissomia.
c) Monossomia e triploidia.
d) Haploidia e triploidia.
e) Euploidia e aneuploidia.
1) C 2) B 3) Gametas anômalos devido à não-disjunção na meiose.
4) E 5) D 6) C 7) C 8) D 9) A 10) C
Nenhum comentário:
Postar um comentário